天价“孤儿药”纳入医保费用直降90%,这些长泡泡的孩子命运等待逆转!
原创:王慧明39健康网2025-05-19 11:31:25
5月17日是世界神经纤维瘤病关爱日,公开数据显示,全球新生儿Ⅰ型神经纤维瘤病的发病率约为1/3000,这意味着在中国至少有几十万名患者,这不是一个小数目。
“我不想交朋友,因为他们说我长得像怪物,不要和我玩……”
“不认识的叔叔阿姨说,我身上的瘤子会传染,要离我远远的……”
“爸爸妈妈带我去了好多医院,有的医生说找不出病因,有的医生说没有办法治疗,要我们回家……”
因为皮肤上出现大大小小的瘤体,远远看上去就像长了一串泡泡,他们被大家称呼为“泡泡宝贝”。
而从出现症状到确诊,“泡泡宝贝”李墨(化名)用了六个月才找到答案,桐桐(化名)则花了四年。
其实他们背后患上的是一种名为神经纤维瘤病的罕见病,是一种由于基因突变而导致的神经系统遗传性肿瘤。临床上分为Ⅰ型神经纤维瘤病(NF1)、Ⅱ型神经纤维瘤病(NF2)和神经鞘瘤病(SN)。其中,Ⅰ型神经纤维瘤病患者的比例占到90%左右。
5月17日是世界神经纤维瘤病关爱日,公开数据显示,全球新生儿Ⅰ型神经纤维瘤病的发病率约为1/3000,这意味着在中国至少有几十万名患者,这不是一个小数目。
中山大学附属第一医院儿科副主任医师徐玲玲教授告诉39深呼吸,她接触的大多神经纤维瘤病患儿都曾因病情导致的外形变化而受到不公的对待。
更令人难过的是,她所在的科室很早就开始接诊这类病人,但由于临床上一直没有有效的治疗手段,导致早期诊断、系统管理和合理治疗等方面面临着较大挑战。“甚至在更早之前,这个病基本无药可治,家长无奈只能带患儿回家观察。”
39深呼吸了解,直至2020年,国际上首款治疗I型神经纤维瘤病的药物才出现。同年,我国首个NF1多中心治疗协作组成立,随后这一模式在全国各地推广,2023年,由中国罕见病联盟、神经纤维瘤病多学科协作组制定的首部《Ⅰ型神经纤维瘤病多学科协作指南》才正式发布。

认识这种病,迈出战胜NF1的第一步
神经纤维瘤病是一类常染色体显性遗传性疾病。
徐玲玲教授介绍,“I型神经纤维瘤病(NF1)最明显的症状就是孩子出生时身体多处有咖啡牛奶斑,或者腹股沟、腋窝出现一些雀斑。随着孩子的生长发育,骨头会受到损害,后期身上的斑块开始变深,还会在皮肤上凸起一个个小包块(结节),俗称‘肉疙瘩’。”
此外,除了表面上的症状外,有1/3的患者会出现丛状的神经纤维瘤。这种良性肿瘤会沿着中枢神经系统、骨骼、脉管系统、内分泌系统等多个系统生长,无孔不入是它最大的特点,全身凡是神经所能触及的地方,都将成为它生长的沃土。
有资料统计,NF1患者瘤体增长不可控,每年患儿肿瘤体积增长15.9%。其中年幼患儿的进展速度更快,3-5岁患儿增长35.1%,8%-13%的患者会发生恶变,恶变后五年总生存率不超过50%,亦会造成逐渐加重的疼痛、毁容、畸形、功能障碍,甚至危及生命。
在临床中,NF1症状的复杂性也给疾病的诊断带来了挑战。徐玲玲教授向39深呼吸介绍,“如果是儿童期,患儿可能会先到儿科就诊,但如果已经到了成年,首发症状为咖啡牛奶斑,患者可能在皮肤科就诊;如果首发症状是皮下或者皮肤上小结节,患者可能在外科就诊。”
这也就意味着容易出现误诊、漏诊的现象。
专家们发现,神经纤维瘤病的患者往往需经新生儿科、影像科、神经科、皮肤科、肿瘤科等多学科联合诊疗后才能得到正确的诊断,而多学科合作是提高神经纤维瘤病的患者治疗水平、生存质量及改善疾病预后等的关键手段。
2020年,我国首个NF1多中心治疗协作组成立,之后这一模式在全国各地推广。目前已有50多家医院启动或成立了神经纤维瘤病相关的多学科门诊和神经纤维瘤病专病门诊。随后,《Ⅰ型神经纤维瘤病多学科协作指南》出炉。
在这版指南里对 NF1 的诊断标准进行了规范:
(1)6 个或以上咖啡牛奶斑:在青春期前直径 > 5 mm 或在青春期后直径 > 15 mm;
(2)2 个或以上任何类型的神经纤维瘤或 1 个丛状神经纤维瘤;
(3)腋窝,腹沟或皮肤褶皱区的雀斑样痣;
(4)视神经胶质瘤;
(5)裂隙灯检查到 2 个或以上 Lisch 结节(虹膜错构瘤),或光学相干层析成像(OCT)/近红外(NIR)影像检查到 2 个或以上的脉络膜异常;
(6)特征性骨病变,如蝶骨发育不良、胫骨前外侧弯曲,或长骨假关节生成;
(7)在正常组织(如白细胞)中具有等位基因变体分数达 50% 的致病杂合子 NF1 变异体。
*对于无父母患病史者,满足 2 条或以上临床特征可被诊断为 NF1;有父母患病史者,满足 1 条或以上临床特征可被诊断为 NF1。
同时 NF1 疾病具有较高的遗传性,约 50% 的 NF1 患者遗传自父亲或母亲,所以做好产前筛查以及新生儿的检查就尤为重要。对于高度怀疑的患者,就一定要做核磁共振检查。
“这几年业界对神经纤维瘤病的认识明显提高了。”徐玲玲教授能感受得到。
压力大:面对周围人异样的目光、歧视和孤立
20岁的上海人李墨是在2020年疫情期间确诊为I型神经纤维瘤病。
“其实小时候身上就出现了咖啡牛奶斑,但穿着衣服旁人也看不到,所以一直没有太在意。”李墨回忆,除了皮肤上的异样,日常就是体力跟不上同龄人。“自己喜欢足球,但体力差上不了场,所以一直被同学们取笑。情绪低落时,我就会拿喜欢的上海申花足球队鼓励自己,他们从低谷慢慢重新复苏,我也一样可以。”
2020年下半年,李墨的身上的咖啡斑变大,甚至出现了凸起的结节,他意识到了危险,在上海九院做了基因检测,查出了病因。
“心里的大石头终于落下来了,但同时我也很绝望。”这是他第一次听说这个病,从医生的口中得知没有根治的方法。“幸运的是入组了当时医院的临床试验组,用上了已经在美国上市的新药。”李墨说,为了能以后能持续用药,他最后选择去香港上大学。

相比于李墨,13岁的桐桐从出生就发现身上十几块大小不等咖啡斑,到六岁因为长出皮下结节,桐桐才被确诊。桐桐的父母说,“最难过的是没有办法治疗。”
在做了核磁共振后,桐桐身体里面发现颈椎、腰椎、胸椎和脊椎旁长满了葡萄串似的肿瘤,最严重的肿瘤生长在口腔中,必须进行手术切除。
不断生长的肿瘤可以用手术切除,但疾病带来的心理创伤还需要很长时间抚平。随着桐桐的慢慢长大,一向乐观也开始变得敏感、焦虑。据其父母回忆,有一次桐桐告诉她们,虽然她告知了同学自己生病,但却在私底下听到同学议论她的长相,说不要和她玩,这让她非常难过。
徐玲玲教授告诉39深呼吸,她在门诊中听到过很多这样的故事,罕见病领域面对的是社会弱势群体,患者能否受到社会的公平对待,也是公民素养和社会发展程度的重要标志之一。
神经纤维瘤病I型患者可能在面部和身体的不同部位长出肿瘤和牛奶咖啡斑,带来容貌和其他身体外观上的损害,而由于公众对疾病的不知道、不理解,使得患者往往会受到公众异样的目光、歧视和孤立。对于一些一出生就伴随着疾病症状的儿童患者来说,这种精神压力陪伴着他们从成长,到就学、就业,到生活和社交,产生了极大的心理负担。
用药难、保障难仍是罕见病患者全体跨不过去的坎
“我希望神经纤维瘤药物能够尽快获批成人适应症。”
这是20岁李墨最大的梦想。他所说的神经纤维瘤药物“司美替尼”,被称作“孤儿药”。2023年,首个针对I型神经纤维瘤病儿童患者的药物在中国上市,且成功纳入医保乙类目录。
包括李墨在内的神经纤维瘤患者,在某种程度上算是幸运儿。当前已知的罕见病中,有90%属于严重疾病,但仅不到5%有治疗方案。
不过,神经纤维瘤虽然在创新疗法可及上已经迈出了重要的一步,然而对于众多罕见病患者及其家庭而言,即便盼来了新药,进入了医保,但 “进院难”、“用药难”、“自付高”的“最后一公里”困境依然亟待解决。
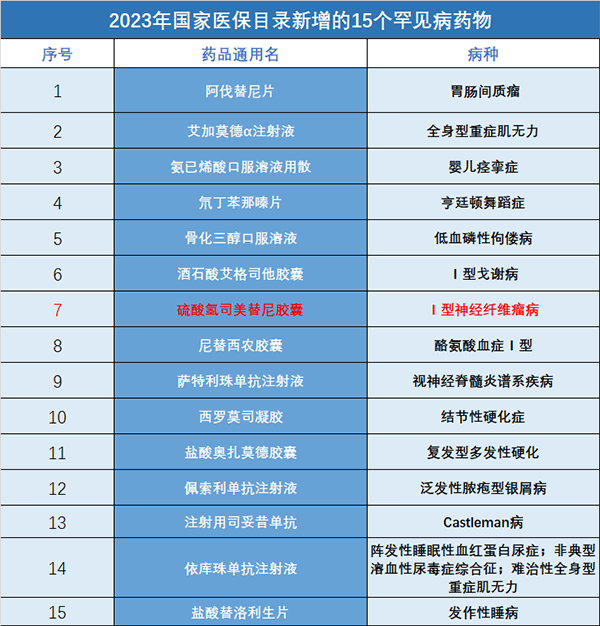
◎ 2023年国家医保目录中,15种罕见病药物被纳入其中,创近三年新高。/ 图:39深呼吸
“药物起效时,我能感觉到肿瘤在缩小,就像春天积雪消融。”李墨作为国内首批使用司美替尼的患者成功实现了在绿荫场上奔跑;这款一盒上万元的“天价药”进入医保后,桐桐很快用上了“救命药”。徐玲玲教授介绍,目前神经纤维瘤患儿门诊用药报销比例以及年龄限制都有区别,例如广东某些地区在政策加持下,经医保支付及大病报销后,个人负担最高可减到90%以上。但也有其它地方的报销比例不足50%,对患儿家庭而言,还是有不小的负担。
中国罕见病联盟神经纤维瘤病专业委员会主任委员、首都医科大学附属北京天坛医院主任医师刘丕楠教授在接受媒体采访时也曾表示,希望今后有更多社会力量关注神经纤维瘤群体面临的巨大挑战,保险、公益机构、慈善力量等各方共同参与,构建多层次保障体系。
面对如何跑赢罕见病“最后一公里”?国家卫健委药物与卫生技术综合评估中心研究员赵琨表示,单一的医疗保障模式难以满足患者的多样化需求,涉及国家、地方、公益慈善、普惠险、商业保险等多层次保障成为重要策略,近年来,惠民保等商业保险对罕见病的保障就发挥着越来越重要的作用。”
在全球7000多种罕见病中,神经纤维瘤群体正站在希望的临界点。他们的故事提醒我们:每个被叫做"泡泡宝贝"的生命,都值得被认真对待;每次对罕见病的关注,都在推动文明向前一步。