上班累到手脚无力,新冠后更严重,没想到得了跟京东副总裁一样的病
原创:葵葵39健康网2024-03-06 15:15:05
罕见病群体是一个庞大的群体,据估计,仅中国的罕见病患者已经超过2000万。还有很多尚未确诊的潜在患者,实际患病人数远不止于这个数。

数据显示,全球目前已知的罕见病超过7000种,中国已知的罕见病大约有1400种。2018年我国公布了《第一批罕见病目录》,共收录121个病种,有40多种与神经系统相关,其中20多种以神经系统受累为主要表现。
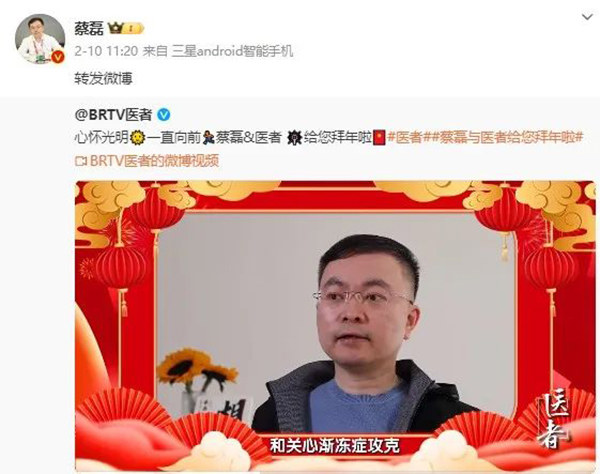
◎ 京东原副总裁蔡磊罹患的渐冻症,就是一种神经系统罕见病,目前无药可治。/图:微博截图
“罕见的少”只是一个相对概念,罕见病的病种纷繁复杂,具体到单个病的患者数量很少,但从医生的直观感受来看,罕见病在临床上并不罕见。
中山大学孙逸仙纪念医院神经科副主任医师何蕾,从事神经系统疾病诊疗13年整,她说,渐冻症、多系统萎缩、自身免疫性脑炎、全身型重症肌无力、视神经脊髓炎、脊髓小脑性共济失调等在神经科的日常诊疗中是比较常见的,只是后来它们被纳入了国家罕见病目录里面。

◎ 何蕾在看诊。(医生供图)
客观的数据亦强烈提示,罕见病群体是一个庞大的群体,据估计,仅中国的罕见病患者已经超过2000万。还有很多尚未确诊的潜在患者,实际患病人数远不止于这个数。
01男子手脚无力,新冠感染后加重,一查竟是渐冻症
2014年 “冰桶挑战计划”风靡全球,被世界卫生组织列为世界五大绝症之首的“渐冻症”,由此进入公众的视野。
著名物理学家霍金、人民英雄勋章获得者张定宇、京东原副总裁蔡磊,都罹患该病。蔡磊更是以自身为武器,和渐冻症抗争到底,他建立了全世界最大的渐冻症患者科研平台,捐资千万发起公益资助计划,令无数人动容。
◎ 蔡磊再捐1亿用于渐冻症研究。微博截图
渐冻症,又称为肌萎缩侧索硬化症(amyotrophic lateral sclerosis,ALS),属于运动神经元病,患者发病时会累及全身的运动神经元,病人的身体开始不受控制,肌肉进行性萎缩无力,通常从手指开始,扩展至躯干和颈部,再发展到吞咽困难、声音嘶哑、呼吸困难、缺氧等,就像逐渐被冰冻住一样。患者又称“渐冻人”。
中国有多少渐冻人?没有一个确切的数据,但根据北京大学第三医院神经内科樊东升教授团队对2012年至2016年中国4亿多医保数据进行的统计调查,我国10万人中每年约有2.97人患渐冻症,1.62人发病。
随着渐冻症越来越被大众熟悉,何蕾在门诊频频遇到疑病患者。“咨询者来自各个年龄段,往往是四肢出现肌肉不受控制地、自发地‘跳动’,或者出现手脚的麻木,病人就会害怕担心是渐冻症。”她解释说, “肉跳”是渐冻症特征性表现之一,但肉跳不一定就是渐冻症,有时候睡眠不好、过度劳累或者精神比较紧张时,都会出现“肉跳”。在完整的病史询问和详细的神经系统查体后,多数病人属于虚惊一场,与渐冻症毫无关系。
但也有少数者不幸确诊了,38岁的张希(化名)是其中之一。

◎ 神经科门诊。/图:作者摄
有一段时间,张希感觉工作特别累,累到力不从心,然后手脚无力,起初以为只是过劳,通过休息就能恢复。不巧又赶上了新冠感染,他“阳康”后手脚无力症状明显加重,且越发严重。不到半年的功夫,张希走路都困难,辗转深圳、东莞等地迟迟得不到确诊。
待找到何蕾看诊时,张希已要坐轮椅了,查体、完善肌电图等检查,他的情况非常符合运动神经元病,随后的全外显子测序结果提示他携带了渐冻症易感基因。
此前不是没有怀疑过,也有医生善意提醒“渐冻症”,但张希与蔡磊一样正值盛年,都是家里的顶梁柱,难以接受这样的事实,反复看诊从医生得到相同的答案,他就不再逃避而是勇敢面对了,在全家人的支持和各种康复锻炼中努力维持着生活质量。
至今,渐冻症被发现近200年的时间,尚未有明确的治愈解决方案,全球只有4款药物上市,且效果微乎其微。从孙逸仙纪念医院出院后,张希又去北京、上海求治,但病情持续进展。
“他最近一次联系我说,吃饭都困难,呛咳厉害,晚上睡觉要佩戴呼吸机,人消瘦了许多,如果进一步恶化可能要插胃管。”何蕾安慰病人要保持积极的心态,有助于病情控制,这是大实话。目前渐冻症的治疗主要是对症治疗,而精神支撑是最好的良药。
包括张希在内,一张张渐冻人的面孔从何蕾的脑海中掠过,她这些年诊断了不少的渐冻症,“从后期随访看,多数渐冻人大概也就活了5年左右”。作为医生,她非常希望尽自己最大的努力去帮助病人,减缓病情进展。
02医生会诊新发现:老人尿频尿急,不全是前列腺惹的祸
很多人的一个共同疑问:为什么现在罕见病越来越多了?
“不是罕见病突然变多了,而是我们的诊疗技术进步了,包括基因检测、影像学检查、电生理检查、或其他的生化检测,可以更好地辅助医生进行诊断。”何蕾解释说。

◎ 检查检测手段的进步,让越来越多罕见病得到诊断。/图:作者摄
但总体而言,罕见病诊断依然困难,患者的症状表现千差万别,而神经系统相关疾病的诊断,被认为是难度最大的,很多病人家属走到她面前一脸愁容说,“我们已经看了很多家医院很多科室,都不知道是什么病,我们现在就想知道是个什么病?”
神经科医生经常被邀请到泌尿外科、骨科进行会诊,从老年尿频尿急或者颈椎病患者中识别出了不少的罕见病多系统萎缩(MSA)患者。
以60岁以上的老人多见,开始是尿频、尿急,半夜频繁起来小便,很多人当前列腺增生症直接去了泌尿外科。但是,治疗了前列腺的问题,又出现了更多的困扰,一起床就头晕,慢慢地走路也没有力气,出现手抖、全身僵硬,而且“行动慢”越来越严重,不敢走路,怕摔跤,最后发现其实是多系统萎缩。
多系统萎缩是一种进展性的神经系统退行性疾病,临床表现为自主神经功能障碍、帕金森综合征和小脑综合征的多种组合。当病情累及自主神经系统,就会影响小便,还会出现性功能障碍;进展到小脑系统,会出现走不稳、头晕,进展到椎体外系统的话,它就会出现身体僵硬、行动慢等一系列的问题。
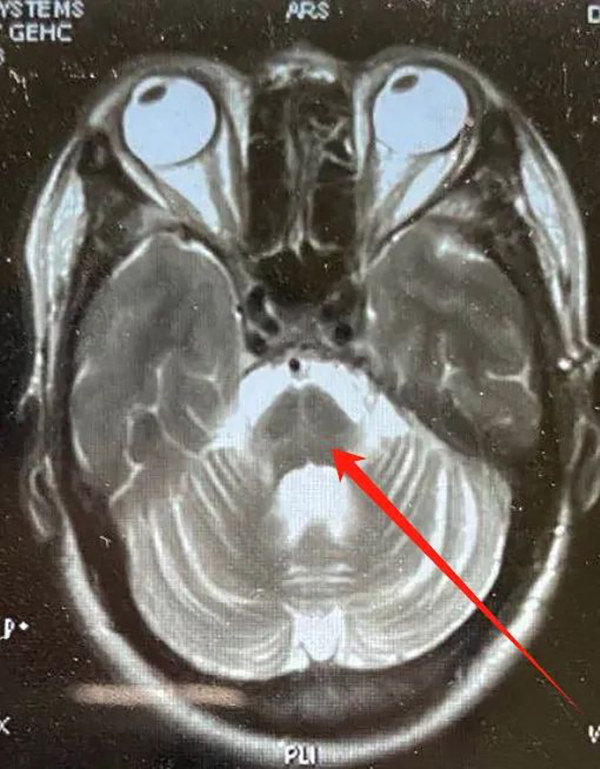
◎ 多系统萎缩患者,影像检查上出现桥脑“十字征”,提示已有脑萎缩 (医生供图)
这些早期症状不典型,非常具有迷惑性,容易被当成人体衰老的正常现象,骨科和泌尿外科都会经常碰到这类老年病人,有经验的外科医生会在门诊甚至手术前通知神经科医生进行评估,排除神经系统病变,避免过度治疗、无效的治疗。
何蕾表示,神经科一年都能碰上数十例多系统萎缩的患者,她也不断在骨科、泌尿外科会诊到这类病人。
在这几年,不少医院推出了罕见病专病门诊,如果遇到疾病诊断不清楚或者治疗效果不好,特别是出现更多的难以解释的症状,建议到罕见病门诊看看。
03体弱男生被父亲怼“偷懒”,没想到孩子病得不轻
治疗比诊断更加难,缺乏特效药,不乏悲观者否定罕见病诊断的价值。何蕾表示不认同,她说,罕见病不完全等同于没得治的遗传病(绝症),有些罕见病是可以治疗的,就算暂无特效药,诊断清楚了,起码在黑暗中有了方向,对病人和家属都有很大意义。

◎ 80%的罕见病与遗传相关。/图:作者摄
“明确的诊断,将改变患者的一生,使之得到恰当的照料,生存质量得到改善。”让何蕾说起了几年前遇到的来自海南的14岁男孩沐沐,她印象非常深刻的一线粒体脑肌病。
沐沐从小不爱动,是一个特别安静的男孩子,妈妈一直以为他是性格使然,然后在沐沐逐渐成长的过程中,就发现他体质很弱,饭量很小,不喜欢上体育课。爸爸是一位典型的中国传统父亲,认为男孩必须阳刚有精神,所以看到儿子成天一副懒洋洋的模样,气就不打一处来。从小学到中学,沐沐没少挨爸爸的责骂,“偷懒”、“没出息”。
直至一次高烧,沐沐全身抽搐,辗转就诊才揭开了他体弱文静的谜底。
妈妈跟医生描述儿子第一次发病的状况:一开始就是严重发烧,然后四肢抽搐,孩子看东西模糊,还很烦躁。在当地医院就诊,怀疑颅内感染,但治疗后病情非但没有好转,反而更加严重,父母收到了医生的病重通知书。
到底是什么感染?棘手的病情,但始终没有让这家人往太坏的地方去想,只当一个正常的孩子偶然生了重病。但何蕾接诊后经过细致的观察发现并不是简单颅内感染,随后的一系列检查明确,沐沐患了一种叫做线粒体脑肌病的罕见病,调整了治疗方案,孩子终于转危为安。
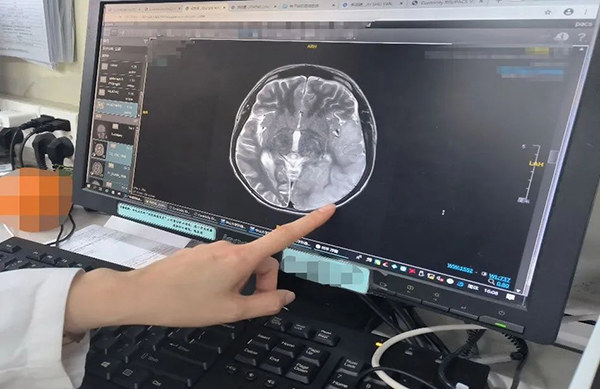
◎ 医生手指的地方提示,沐沐的大脑皮层肿胀。/图:作者摄
“这个孩子从小到大都不爱动,体力不好,用这个病也就解释得通了。”听了医生的一番解释,沐沐的妈妈泣不成声,“原来我们一直都错怪了孩子,他不是真的懒……”
线粒体脑肌病是一种遗传性疾病,包括最常见的母系遗传以及常染色体显性遗传、常染色体隐性遗传等。中山大学孙逸仙医院神经科自1971年成立至今,收治了多例线粒体脑肌病患者。
该病需要通过饮食、代谢治疗和对症治疗来综合管理,且这类病人的用药要很小心,影响线粒体功能的药物都要比较慎用,比如降血脂的他汀类药物、双胍类降糖药,围手术期更加要注意电解质稳定和能量的补充;虽然病人也有癫痫发作,但抗癫痫药的选择方面也要很慎重,不同于单纯的癫痫患者。

◎ 患有罕见病,日常用药要很小心。/图:作者摄
在饮食起居上,患者的一日三餐要注意,应当保持充足的饮食以维持能量代谢的平衡和稳定,避免饥饿、饮酒、高脂肪低糖饮食。有氧耐力锻炼可以提高患者的肌力,但应适度,不能过度劳累。
而家里人自从知道沐沐的病因,在生活照料上更加用心。经过几年的治疗和调养,沐沐的病情控制稳定,他长高长胖了,还能够像同龄人一样正常上学,但不会有同龄人的来自父母的压力,今年就要高中毕业了。
罕见病孩子基本能够正常的生活,就是对父母最大的安慰。如果没有那一份确诊书,沐沐可能还一直活在“懒孩子”的阴影中。
(文中患者和家属均为化名)
通讯员|黄睿